Base Repo : wings-public
Homepage : wings-platform.org
Introduction
This tutorial will guide you through the process of querying federated genomics data hosted in WiNGS, using python based access to the REST api. We’ll cover the following sections:
- Understanding Genetic Data and VCF Files
- Setting up the python conda environment
- Acquiring an api key
- Querying data
- Manipulating data
google colab page for this demo
1. Understanding Genetic Data, VCF Files and the federated setup of WiNGS
VCF files
Genetic data is usually provided in Variant Call Format (VCF) files. These files contain information about genetic variants, including the position, reference, and alternate alleles, as well as additional annotations. VCF files are commonly used in genomic research to store and share genetic variant data. It is important to keep in mind that over time, multiple versions of ‘the human genome’ have been released, and variant positions might shift slightly between these releases.
VCF files can contain data from a single sample, or from multiple samples. In the latter case, samples are organized is columns, holding information for each encountered position. Although these files can be filtered and analyzed using multiple tools such as vcftools or pyVCF, adding more data over time would require to rebuild them iteratively. Another limitation is formed by the privacy constraints on genomic data, prohibiting the collection of cross-center cohorts for large-scale statistical analysis.
Principle of WiNGS
For these reasons, WiNGS was implemented to become a portal to aggregated genomics data, kept privately in a federated framework. WiNGS supports data in multiple of the human reference versions (or genome builds) and provides unified, biological annotations. This way, variants in all federated data hubs, will have uniform annotations (or meta data).
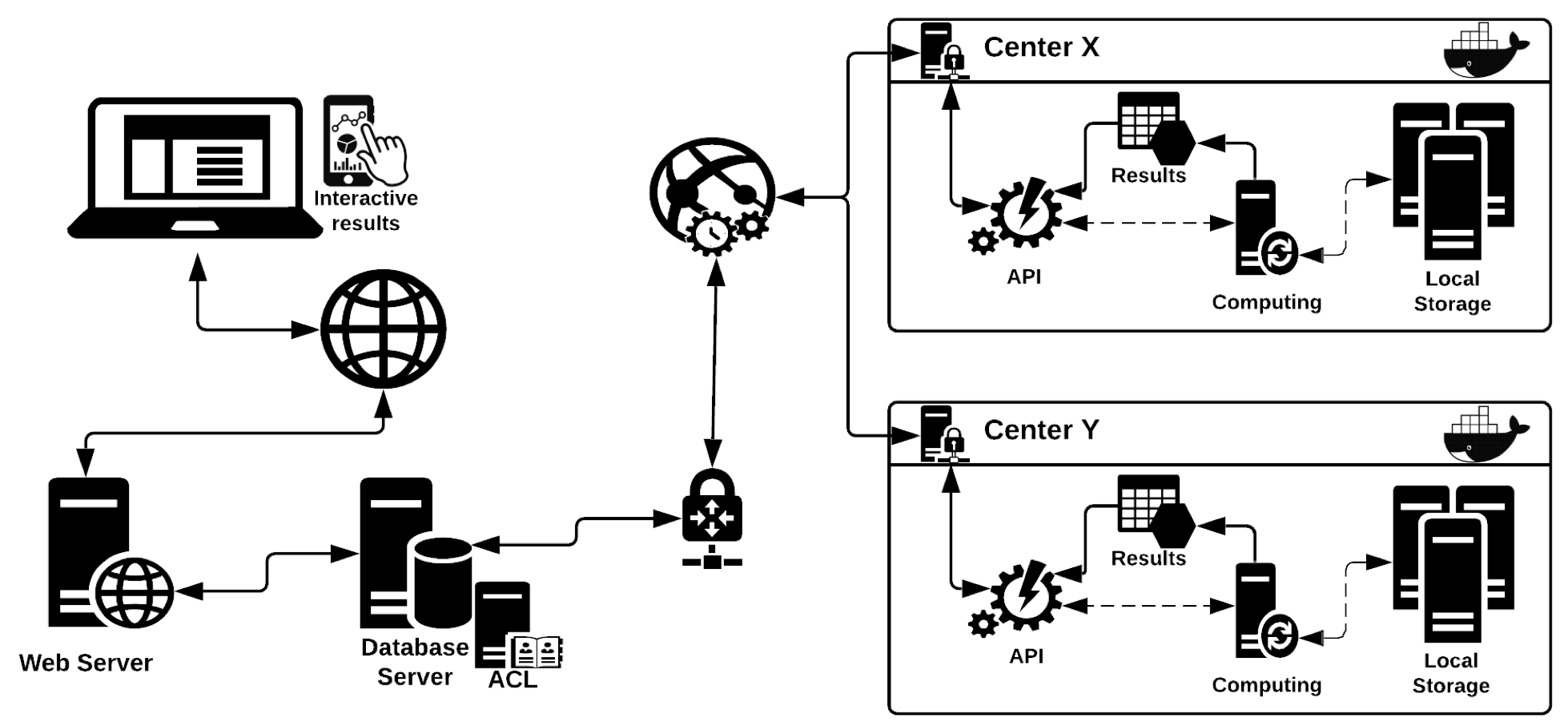
Data is structured in WiNGS as follows:
- ** Individual **: these are cinical cases, patients.
- ** Sample **: these are the experimental samples associated to individuals. (eg : WGS experiment, or SV experiment)
- ** Dataset **: These are the analysis files related to a sample. eg : GATK based SNV analysis on hg38 for WGS data.
WiNGS is accessible as REST api, which is documented through swagger. To perform an analysis, the following prerequisites are needed, which are performed through a web-based UI:
- ** A user account ** : to shield data, anonymous access is not allowed, register here
- ** An API key ** : Log in and select user meny : Create API key , on the top right
2. Setup conda environment
Install miniconda
We use miniconda for this tutorial. Follow the Installation Instructions. For example on linux:
mkdir -p ~/miniconda3
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh -O ~/miniconda3/miniconda.sh
bash ~/miniconda3/miniconda.sh -b -u -p ~/miniconda3
rm ~/miniconda3/miniconda.sh
Install the prerequisites :
conda create -n wings python=3.10 pandas xlsxwriter
conda activate wings
3. Test Connection
Save the following script as WiNGS_api.py
#!/usr/bin/env python
@title IMPORTS
import getpass
import requests
import time
from tqdm import tqdm
import pprint
import pandas as pd
import matplotlib.pyplot as plt
import hashlib
import json
%matplotlib inline
@title GENERAL CLASSES
#############
## CLASSES ##
#############
class APIError(ValueError):
def __init__(self, response, value: dict):
self.response = response
self.value : dict = value
class WingsApi:
SLEEP_SECONDS = 5
RETRY = 60 # number of times to retry, wait SLEEP seconds after each failure
def __init__(self,url,token):
self.url = url.rstrip('/')
self.token = token
self._job_cache = {}
self.session = requests.session()
self.session.headers.update({'Authorization': f'Bearer {self.token}'})
self.session.headers.update({'Content-Type': 'application/json'})
def hash_request(self,data):
# Convert dictionary to a JSON string with sorted keys to ensure consistent ordering
json_str = json.dumps(data, sort_keys=True)
# Create an MD5 hash object
md5_hash = hashlib.md5()
# hash the json
md5_hash.update(json_str.encode('utf-8'))
# Get the hexadecimal representation of the hash
return md5_hash.hexdigest()
def get(self, endpoint, arguments={},skip_cache=False):
md5 = self.hash_request([endpoint, arguments])
if md5 in self._job_cache and not skip_cache:
return self._job_cache[md5]
else:
r = self.session.get(f"{self.url}/{endpoint}",params=arguments)
if r.status_code != 200:
raise APIError(r,r.json())
self._job_cache[md5] = r.json()['message']
return r.json()['message']
def post(self,endpoint,arguments={},skip_cache=False):
md5 = self.hash_request([endpoint, arguments])
if md5 in self._job_cache and not skip_cache:
return self._job_cache[md5]
else:
r = self.session.post(f"{self.url}/{endpoint}",json=arguments)
if r.status_code != 200:
raise APIError(r,r.json())
self._job_cache[md5] = r.json()['message']
return r.json()['message']
def individual_details(self,individual):
# clinical data
endpoint=f"individuals/{individual['IndividualID']}"
r = self.get(endpoint)
individual['phenotype'] = r[0]['phenotype']
# trio's defined ?
endpoint='families'
r = self.get(endpoint,{'proband' : individual['IndividualID']})
if len(r):
individual['family'] = r
endpoint = 'trios'
r = self.get(endpoint,{'family' : individual['family']['FamilyID']})
r = [x for x in r if x['TrioStatus'] != 'disabled']
if len(r):
individual['trio'] = r
else:
individual['trio'] = None
else:
individual['family'] = None
individual['trio'] = None
# samples (data)
endpoint='samples'
r = self.get(endpoint,{'host_id' : 1, 'piid' : individual['PIID']})
r = [x for x in r if x['IndividualID'] == individual['IndividualID']]
if len(r):
individual['samples'] = r
else:
individual['samples'] = r
return individual
def get_results(self,endpoint,arguments,post=False):
results = list()
# first call : with cache
if post:
r = self.post(endpoint,arguments)
else:
r = self.get(endpoint,arguments)
if r.get('status') == 'ready':
results.extend(r['results'])
# pages ?
page = 1
while r.get('meta').get('last_page') == 'false':
page += 1
#print(f"Fetching page {page}...")
arguments['page'] = page
if post:
r = self.post(endpoint,arguments)
else:
r = self.get(endpoint,arguments)
results.extend(r['results'])
return results
# not ready / cached: poll status
for _ in tqdm(range(self.RETRY)):
r = api.get(endpoint,arguments,skip_cache=True)
status = r.get("status")
if status == "not ready":
time.sleep(self.SLEEP_SECONDS)
continue
elif status == "ready":
page = 1
results.extend(r['results'])
while r.get('meta').get('last_page') == 'false':
page += 1
print(f"Fetching page {page}...")
arguments['page'] = page
if post:
r = self.post(endpoint,arguments)
else:
r = self.get(endpoint,arguments)
results.extend(r['results'])
return results
raise ValueError(r)
print(f"\nJOB: still in progress. Stop polling... ")
return None
def get_variant_results(self,endpoint,arguments):
results = list()
# first call : with cache
r = self.get(endpoint,arguments)
if r.get('status') in ['completed',"no-variants"]:
return r
# not ready / cached: poll status
for _ in tqdm(range(self.RETRY)):
r = self.get(endpoint,arguments,skip_cache=True)
status = r.get("status")
if status == "inprogress":
time.sleep(self.SLEEP_SECONDS)
continue
elif status in ["completed","no-variants"] :
return r
raise ValueError(r)
print(f"\nJOB: still in progress. Stop polling... ")
return None
if __name__ == '__main__':
# provide your token :
token = getpass.getpass('Enter your token : ')
# the url of the api :
url="https://wings.esat.kuleuven.be/rest-api/"
# setup the api class
api = WingsApi(url,token)
# get the principle investigator ID (piid) associated to your user
endpoint='piid'
r = get_api_result(url,endpoint,token)
print(f"The resulting data is : \n {pprint.pformat(r)}")
# select first (if multiple)
piid = r[0]['PIID']
When executed, the above script will ask for your token and will print the api response to screen. Note the piid as you will need the value later on.
4. Listing Samples
Samples access is restricted by account. This means that as a registered user, you can only see samples assigned to you. This type of work is not federated in a strict sense, as WiNGS automatically directs queries to the correct, individual data node. We’ll move on to cross-node routines in later sections.
For now, we’ll investigate what samples you have access to, select a family (inddex + parents) and perform variant querying on them.
Add the following snippet to the main section of the script:
# list all individuals
endpoint='individuals'
r = api.get(endpoint)
print(f"You have access to {len(r)} individuals:")
individual = None
# input for matching the individual you want :
#s_string = input('Provide the LocalID of the individual to select: ')
s_string = "Demo_index"
for idx, i in enumerate(r):
if i['LocalID'] == s_string:
individual = i
if idx < 5:
print(f"{i['LocalID']} : {i['IndividualID']}")
if idx == 5:
print('....')
if not individual:
raise Exception('No Individual found')
It will list all individuals and then select “Demo_index” as an example. It is part of a public dataset all users have access to.
Information about the individual of interest
The following code will fetch information about the family members linked to the sample and set phenotypes.
# get more details, see class for details on endpoints used
individual = api.individual_details(individual)
print("\nDetails on the selected individual : ")
pprint.pprint(individual)
# get variables for use in querying :
used_trio = individual['trio'][0]
used_sample = [x for x in individual['samples'] if x['SampleFileID'] == used_trio['ProbandFileID']][0]
The information printed includes phenotypes (HPO) and family members. If family members, with data, are assigned, a TRIO will be created as well. Trio’s contain precalculated in heritance patterns of all variants in the proband, which can be used to speed up filtering.
We selected the first available trio in the list. In case there are multiple datafiles (WES, WGS, …) , multiple trio’s an exists.
5. Querying Data : Sample Based Analysis
General querying
As a first analysis, it might be interesting to look at general variant characteristics in the data. For this, we run a broad filter to obtain all exonic variants:
# first locate the filter
filter = "Statistics_filter"
r = api.get('samples/filter')
r = [x for x in r if x['FilterName'] == filter]
if len(r):
filter = r[0]
else:
raise Exception('Specified filter not found')
# Filter details:
r = api.post('samples/filter/leaf',{'filter_id':filter['FilterID']})
print(f"\nAvailable filter leafs in selected filter: {filter['FilterName']}")
pprint.pprint(r)
print("\n => Specify your selection as comma-separated string to 'filter_level'")
# we take the pass (exonic effects) side:
filter_level = "2"
This code returns all precomposed filters, from which a filter named “Statistics_filter” is retained. Next, we decide what leafs in the filter-tree we want to use. This tree is composed of different yes/no criteria which are hierarchicaly defined. Next, we launch the query:
# submit
print(f"\nLaunching filter with details: ")
pprint.pprint(arguments)
r = api.post(endpoint,arguments)
query_id = r['request_id']
print(f"\nLaunched query as ID : {query_id}")
# wait for results
used_sample_name = used_sample['SampleLocalID']
pprint.pprint({'request_id':query_id, 'local_id' : used_sample_name})
variants = api.get_results("/samples/discovery/results",{'request_id':query_id, 'local_id' : used_sample_name})
print(f"\nNr.Variants : {len(variants)}")
In case of the demo sample, this results in 52K variants. We can plot these according to location, or plot average variant coverage by chromosome:
# working with variants: some stats
df = pd.DataFrame(variants)
# subset df: keep chr, type, phred/cadd and some other stuff.
df = df[['_id','v_type','vcf_chr', 'alt_cnt', 'ref_depth','alt_depth', 'mapping_quality','delta_pl', 'phred_score']]
df['total_depth'] = df['ref_depth'] + df['alt_depth']
# per chr
counts_per_chr = df.groupby("vcf_chr").size().reindex(df['vcf_chr'].unique())
plt.figure()
counts_per_chr.plot(kind='bar', title='Variants per Chromosome')
plt.gca().spines[['top', 'right',]].set_visible(False)
plt.show()
# avg total depth by chr:
avg_total_depth_by_chr = df.groupby('vcf_chr')['total_depth'].mean()
plt.figure()
avg_total_depth_by_chr.plot(kind='bar', title='Average Total Depth by Chromosome')
plt.gca().spines[['top', 'right',]].set_visible(False)
plt.show()
Family based querying
In most cases, variants are interpreted in the context of inheritance. WiNGS provides the following inheritance-based filters when trio’s are defined:
- maternal
- paternal
- denovo
- recessive
- compound-heterozygous
Here we demonstrate the usage of the denovo filter:
# list filters available for TRIO analysis:
endpoint='trios/filter'
r = api.get(endpoint)
print(f"The following filters are available:")
for f in r:
print(f"{f['FilterID']} : {f['FilterName']}")
# select filter id for the demo filtering
r = [x for x in r if x['FilterName'] == 'trio analysis usecase filter']
filter = r[0]
# what leafs shall we take:
r = api.post('trios/filter/leaf',{'filter_id':filter['FilterID']})
print(f"\nAvailable filter leafs in selected filter: {filter['FilterName']}")
pprint.pprint(r)
print("\n => We take: '6,8'")
filter_level = '6,8'
# launch
print("\n => Launching DE NOVO analysis")
arguments = {
'filter_id': filter['FilterID'],
'type' : 'denovo',
'trio_local_id' : used_trio['TrioLocalID'],
'filter_level' : filter_level,
}
r = api.post('trios/inheritance',arguments)
query_id = r['request_id']
print(f"\nLaunched query as ID : {query_id}")
# get results
variants = api.get_results("/trios/inheritance/results",{'request_id':query_id, 'trio_local_id': used_trio['TrioLocalID']})
print(f"\nNr.Variants : {len(variants)}")
Representing the results in a table, would require the following code:
# scoring of effects to select highest impact / variant
scores = {'None' : -1,
'upstream_gene_variant' : 0,
'synonymous_variant' : 2,
'downstream_gene_variant' : 0,
'stop_retained_variant' : 1,
'inframe_deletion' : 3,
'stop_lost' : 6,
'frameshift_variant' : 7,
'splice_region_variant' : 3,
'missense_variant' : 4,
'non_coding_transcript_exon_variant' : 1,
'splice_donor_5th_base_variant' : 3,
'splice_polypyrimidine_tract_variant' : 3,
'splice_donor_variant' : 7,
'non_coding_transcript_variant' : 3,
'start_retained_variant' : 1 ,
'coding_sequence_variant' : 1,
'protein_altering_variant' : 3,
'intergenic_variant' : 0,
'stop_gained' : 7,
'inframe_insertion': 3,
'splice_acceptor_variant' : 7,
'intron_variant' : 1,
'start_lost' : 7,
'splice_donor_region_variant' : 3,
'3_prime_UTR_variant' : 1,
'5_prime_UTR_variant' : 1
}
# create a table summarizing the data:
df = pd.DataFrame(variants)
df = df.drop(columns=['fileID','var_validate','var_key','pid','gt_ratio','non_variant','chr','stop_pos','multi_allelic','stretch_lt_a','stretch_lt_b','trio_code','motif_feature_consequences','regulatory_feature_consequences','RNACentral'])
max_genes = []
for i,row in df.iterrows():
genes = row['gene_annotations']
max_impact = 'None'
max_impact_gene = {}
for gene in genes:
for t in gene['consequence_terms']:
if scores[t] > scores[max_impact]:
max_impact = t
max_impact_gene = gene
df.at[i,'gene_annotations'] = "{};geneid:{};impact:{};consequences:{}".format(max_impact_gene['csn'],max_impact_gene['gene_id'],max_impact_gene['impact'],";".join(max_impact_gene['consequence_terms']))
df.at[i,'gnomAD'] = '.' if not row['gnomAD'] else "AF:{};AN:{}".format(round(row['gnomAD']['AF'],3),row['gnomAD']['AN'])
max_genes.append(max_impact_gene)
# print chr, start_pos, ref, alt, ref_depth/alt_depth, gnomad (or None if empty), gene|impact , gene|consequences
print("\nResulting Variants: ")
print(df.to_string(index=False))
For this demonstration sample, we could identify 17 variants, out of which 8 affected a transcript. We’ll investige these further in the next section.
6. Querying Data : Variant/Gene Based Analysis
Variant Based
Interestingly, some of the variants were not present in gnomAD. To investige this is more detail, we can look at the frequency within WiNGS:
# submit query for the variant on chromosome 8
endpoint = 'variant/frequency'
arguments = {"variant": "8-143818425-C-A",
"ref_build_type": "hg38" }
r = api.post(endpoint,arguments)
query_id = r['request_id']
print(f"\nLaunched query as ID : {query_id}")
# get results
variant_info = api.get_variant_results("/variant/frequency/results",{'request_id' : query_id})
#pprint.pprint(variant_info)
# "overall" to df
df = pd.DataFrame(pd.json_normalize(variant_info["overall"]))
# print the df
print(df.to_string(index=False))
This would show us that the variant is not present in 2800+ WES samples, and 3 times in roughly 900 WGS samples.
| exp | ref_build | cnt | sample-size | het | hom-alt |
|---|---|---|---|---|---|
| WGS | hg38 | 2 | 903 | 2 | 0 |
| WGS | hg19 | 1 | 32 | 1 | 0 |
| WES | hg19 | 0 | 2866 | 0 | 0 |
Gene Based
Next, we will investigate if other high impact variants are present in the same gene (PUF60). To do this, we apply a “high impact” filter, using the gene-id as starting point:
# what filter:
endpoint='variant/discovery/filter'
r = api.get(endpoint)
print(f"The following filters are available:")
for f in r:
print(f"{f['FilterID']} : {f['FilterName']}")
print("\n => We use filter 'Potentially_relevant_variants'")
filter = [x for x in r if x['FilterName'] == 'Potentially_relevant_variants'][0]
# what leafs:
r = api.post('variant/discovery/filter/leaf',{'filter_id':filter['FilterID']})
print(f"\nAvailable filter leafs in selected filter: {filter['FilterName']}")
pprint.pprint(r)
print(f"\n => We take '2,4,6'")
filter_level = '2,4,6'
# run filter on WGS data in hg38
wgs_results = {}
wes_results = {}
wgs_result = {}
wes_result ={}
gene = 22827
arguments = {
'filter_id' : filter['FilterID'],
'host_id' : 1,
'ref_build_type' : 'hg38',
'filter_level' : filter_level,
'query_type' : {'geneID' : gene},
'seq_type' : 'WGS',
'hpo_list' : [x['HPOID'] for x in individual['phenotype']]
}
wgs = api.post('variant/discovery/query/',arguments)
print(f" => Launched WGS query as ID : {wgs['request_id']}")
wes = api.post('variant/discovery/query/',arguments)
print(f" => Launched WES query as ID : {wes['request_id']}")
# get results
wgs_variants = api.get_variant_results("/variant/discovery/query/results",{'request_id':wgs['request_id']})
wes_variants = api.get_variant_results("/variant/discovery/query/results",{'request_id':wes['request_id']})
We also provided the phenotype of our demo individual. This allows us to group data based on the presence of these hpo terms. The results are :
| WGS | |||
|---|---|---|---|
| var+phen | var-phen | -var+phen | -var-phen |
| 0 | 2 | 7 | 460 |
| WES | |||
|---|---|---|---|
| var+phen | var-phen | -var+phen | -var-phen |
| 0 | 0 | 7 | 462 |
Note that the original variant is no longer retained (no var+phen entries). This can be explained by the more stringent filtering criteria (CADD >= 30).
When we investigate the gene with regard to phenotypes, we’ll find that it is causative for verhije syndrome omim:615583. The phenotypic desscription of this syndrome is in line with the phenotype of the demo individual.
A final test would be to investigate if the variant is significantly associated to the phenotype. For this, we’ll use the a less stringent filtering to make sure we retain the variant in question.
# what filter:
endpoint='variant/discovery/filter'
r = api.get(endpoint)
print(f"The following filters are available:")
for f in r:
print(f"{f['FilterID']} : {f['FilterName']}")
print("\n => We use filter 'Potenially_relevent_by_cadd'")
filter = [x for x in r if x['FilterName'] == 'Potenially_relevent_by_cadd'][0]
# what leafs:
r = api.post('variant/discovery/filter/leaf',{'filter_id':filter['FilterID']})
print(f"\nAvailable filter leafs in selected filter: {filter['FilterName']}")
pprint.pprint(r)
print(f"\n => We take '2'")
filter_level = '2'
gene = 22827
arguments = {
'filter_id' : filter['FilterID'],
'host_id' : 1,
'ref_build_type' : 'hg38',
'filter_level' : filter_level,
'query_type' : {'geneID' : gene},
'seq_type' : 'WGS',
'hpo_list' : [x['HPOID'] for x in individual['phenotype']]
}
wgs = api.post('variant/discovery/query/',arguments)
arguments['ref_build_type'] = 'hg19'
wes = api.post('variant/discovery/query/',arguments)
print(f" Waiting for results for gene-id {gene}")
wgs_variants = api.get_variant_results("/variant/discovery/query/results",{'request_id':wgs['request_id']})
wes_variants = api.get_variant_results("/variant/discovery/query/results",{'request_id':wes['request_id']})
The results are now:
| WGS | |||
|---|---|---|---|
| var+phen | var-phen | -var+phen | -var-phen |
| 2 | 5 | 10 | 786 |
| WES | |||
|---|---|---|---|
| var+phen | var-phen | -var+phen | -var-phen |
| 1 | 0 | 11 | 791 |
Based on these counts, we can run a fisher exact test:
endoint = 'variant/discovery/statistics'
arguments = {
"overall": {
"var+phen": 3,
"var-phen": 5,
"-var+phen": 21,
"-var-phen": 1577
},
"stats_test": "fisher test"
}
result = api.post(endpoint,arguments)
pprint.pprint(result)
7. Querying Data : Structural Variant
single sample
A second data type, next to SNV variants, are structural variants. Similar to SNVS, single samples can be queried in depth. However, for SVs, the filters are provided as part of the call:
# list all samples with SVS:
endpoint = "SV_samples"
sv_samples = api.get(endpoint,{"piid": piid, "host_id": 1})
pprint.pprint(sv_samples[0:3])
##
arguments = {"host_id" : 1,
"piid" : piid,
"individualID" : sv_samples[0]['IndividualID'],
"fileID" : sv_samples[0]['SampleFileID'],
"sv_type" : "DEL",
"sv_len" : ">1000"}
svs = api.post('SV_samples/discovery',arguments)
query_id = svs['request_id']['process_id']
print(f"\nLaunched query as ID : {query_id}")
# get results
results = api.get_results("/SV_samples/discovery/results",{'request_id':query_id, 'host_id' : 1}, post=True)
pprint.pprint(results)
populations
SV calling on WGS data often results in large amount of variants. A valuable approach is then to exclude variants based on a control population. This is done by using the populations endpoint:
Populations are similar to datasets for SNV, and allow grouping of samples for analysis. Listing your populations is done through the following call:
r = api.get('populations',{'host_id': 1,'piid' : piid})
pprint.pprint(r['message'])
Assuming no populations are defined, we can create one for the demo samples:
# create a population:
r = api.post('newPopulation',{'host_id': 1, 'piid': piid, 'population_name': 'demo_samples', 'description':'demo_population', 'seqType': 'WGS'})
# get the population ID
r = api.get('populations',{'host_id': 1,'piid' : piid},skip_cache=True)
population_id = None
for p in r['message']:
if p['Description'] == 'demo_population':
population_id = p['PopulationID']
break
# then, add samples with "demo" in the IndividualLocalID.
data = {
"host_id": 1,
"piid": piid,
"PopulationID": population_id,
"individualsAndSamples": []
}
for s in sv_samples:
if 'demo' in s['IndividualLocalID'].lower():
data['individualsAndSamples'].append({
"individualID": s['IndividualID'],
"fileID": s['SampleFileID']
})
r = api.post('population/addIndividualSample',data)
Next, we can investigate the effect by repeating the single sample query in context of the population.
endpoint = 'population/populationFileFiltering'
data = {
"PopulationID": population_id,
"fileID_to_filter": sv_samples[0]['SampleFileID'],
"host_id": 1,
"sv_type" : "DEL",
"sv_len" : ">1000"
}
r = api.post(endpoint,data)
request_id = r['request_id']['process_id']
# get results
endpoint = 'population/populationFileFiltering/results'
data = {'host_id': 1, request_id: request_id}
results = api.get_results(endpoint,data, post=True)
pprint.pprint(results)
In this case, we see that the that number of retained variants decreased significantly.
Cross-node queries
Finally, we can also execute cross-node queries, similar to the variant frequency queries in SNV analysis. For instance, we could investigate if the WiNGS populations contain potentially relevant SV events in the PUF60 gene, identified in the previous section. We do not restrict the query on variant size or type, only on the gene and ACMG class:
endpoint = 'SVvariant/discovery/query'
data = {
'host_id' : 1,
'ref_build_type' : 'hg38',
'seq_type' : 'WGS',
'gene' : 'PUF60',
'ACMG_class' : 3,
'hpo_list': [x['HPOID'] for x in individual['phenotype']]
}
r = api.post(endpoint,data)
request_id = r['message']['request_id']
# get results
endpoint = 'SVvariant/discovery/query/results'
data = {'request_id': request_id}
results = api.get_results(endpoint,data, post=True)
pprint.pprint(results)
The results follow the same structure as the variant frequency analysis.
| var+phen | var-phen | -var+phen | -var-phen |
|---|---|---|---|
| 1 | 20 | 15 | 125 |
Conclusion
The WiNGS api offers standardized interaction with federated genomics data. This tutorial has covered the basic usage of the api. For more detailed information, please refer to the documentation.
More information
Contributors
UZA / University of Antwerp